
Tools for systems-level analysis of Molecular Dynamics (MD) simulations
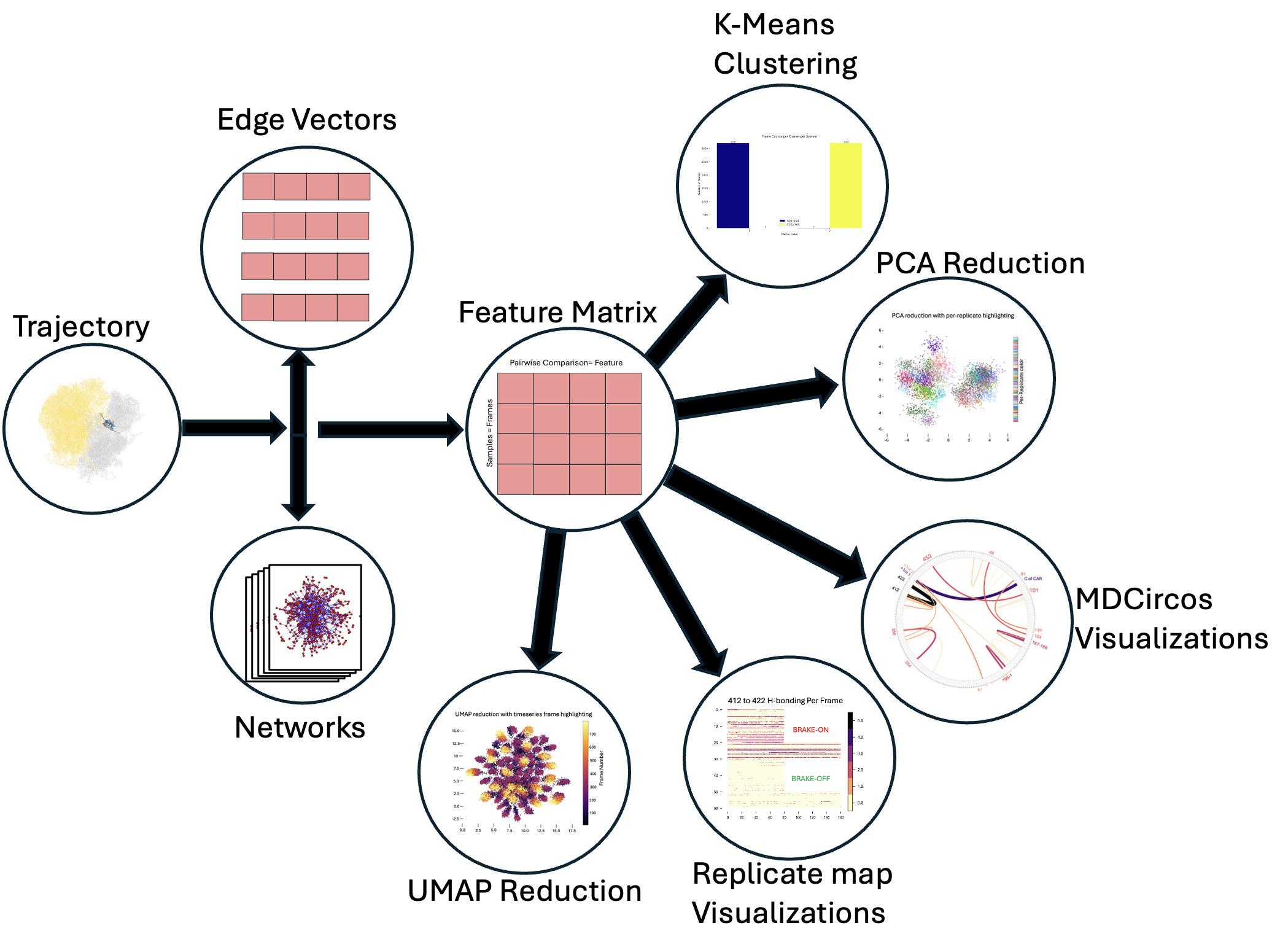
We start from an MD trajectory and generate per-frame interaction networks. We represent theese as edge vectors (vectors consisting of just the edgeweights for every edge connecting pairs of unique nodes); stacking these per-frame vectors yields a feature matrix suitable for clustering (e.g., k-means) and dimensionality reduction (PCA/UMAP). Results can be visualized with graphs, scatter plots, MDcircos plots (residue H-bonding), or replicate maps of frame-level measurements of interest.
pip install mdsa-tools
In the Weir Group at Wesleyan University, we perform molecular dynamics (MD) simulations of a ribosomal subsystem to study tuning of protein translation by the CAR interaction surface — a ribosomal interface identified by the lab that interacts with the +1 codon (poised to enter the ribosome A site). Our "computational genetics" research focuses on modifying adjacent codon identities at the A-site and the +1 positions to model how changes at these sites influence the behavior of the CAR surface and correlate with translation rate variations.
Moving forward the most recent work will be through github releases and PyPI pushes will be the most recent confirmed covered working release.
Quickstart example (see docs for more examples):
from mdsa_tools.Data_gen_hbond import TrajectoryProcessor as tp
import numpy as np
import os
###
### Datagen
###
# load in and test trajectory
system_one_topology = '../PDBs/5JUP_N2_CGU_nowat.prmtop'
system_one_trajectory = '../PDBs/CCU_CGU_10frames.mdcrd'
system_two_topology = '../PDBs/5JUP_N2_GCU_nowat.prmtop'
system_two_trajectory = '../PDBs/CCU_GCU_10frames.mdcrd'
test_trajectory_one = tp(trajectory_path=system_one_trajectory, topology_path=system_one_topology)
test_trajectory_two = tp(trajectory_path=system_two_trajectory, topology_path=system_two_topology)
# now that it's loaded, make objects
test_system_one_ = test_trajectory_one.create_system_representations()
test_system_two_ = test_trajectory_two.create_system_representations()
np.save('test_system_one', test_system_one_)
np.save('test_system_two', test_system_two_)
###
### Analysis
###
from mdsa_tools.Analysis import systems_analysis
all_systems = [test_system_one_, test_system_two_]
Systems_Analyzer = systems_analysis(all_systems)
# transform adjacency matrices, perform clustering and dimensional reduction
Systems_Analyzer.replicates_to_featurematrix()
optimal_k_silhouette_labels, optimal_k_elbow_labels, centers_silhouette, centers_elbow = Systems_Analyzer.perform_kmeans(outfile_path='./test_', max_clusters=5)
print('clustering successfully completed')
X_pca, weights, explained_variance_ratio_ = Systems_Analyzer.reduce_systems_representations(method='PCA') # you could do method='PCA'/'UMAP' here
print('reduction successful')
###
### Visualization
###
import matplotlib.cm as cm
from mdsa_tools.Viz import visualize_reduction
# visualize embedding space with original clusters
visualize_reduction(X_pca, color_mappings=optimal_k_silhouette_labels, savepath='./PCA_', cmap=cm.plasma_r)
# map transitions between various cluster assignments
from mdsa_tools.Viz import replicatemap_from_labels
fake_labels = np.arange(0, 18, 1)
replicatemap_from_labels(cmap=cm.plasma_r, frame_list=[9] * 2, labels=fake_labels, savepath='./Repmap_') # 9 frames each