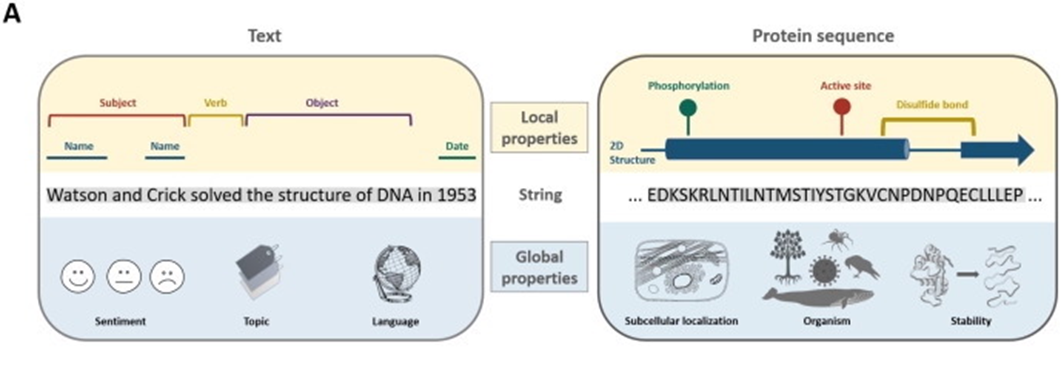
Malaria is a life-threatening disease caused by the Plasmodium parasite transmitted to humans through the bite of infected female Anopheles mosquitoes. The Plasmodium falciparum parasite is particularly deadly and responsible for most malaria-related deaths globally. Proteins play a crucial role in enabling the Plasmodium falciparum parasite to survive within a human host. Biologists use standardized annotation terms such as Gene Ontology (GO) and Protein Families (pFam) to classify and group proteins based on their functions. However, a significant number of proteins in the Plasmodium falciparum proteome have unknown functions, thus hindering our broader understanding of the parasite's survival mechanisms.
Scientists have used sequence similarity to annotate proteins, but this method has limitations, particularly when dealing with highly specific proteins that cannot be found in other species, such as most of the "unknown function" proteins found in Plasmodium falciparum. Fortunately, large protein language models (LMs) trained on manually curated annotated protein databases such as SwissProt have proven successful in annotating proteins.
Our objective is to generate fine-tuned vectorized representations of each protein sequence in the training dataset. We will refine the embedded representations to ensure that proteins sharing the same pFam annotation cluster closely together within the latent space, while those with different pFam terms remain further apart. This refined dataset will serve as our lookup table. We will then encode the proteins with unknown functions (test dataset) and examine the nearest neighbors for each protein sequence in the fine-tuned lookup table. We will evaluate if there is consensus in the annotation of the nearest neighbors and if we can "bridge" the annotation over to the unknown function protein.
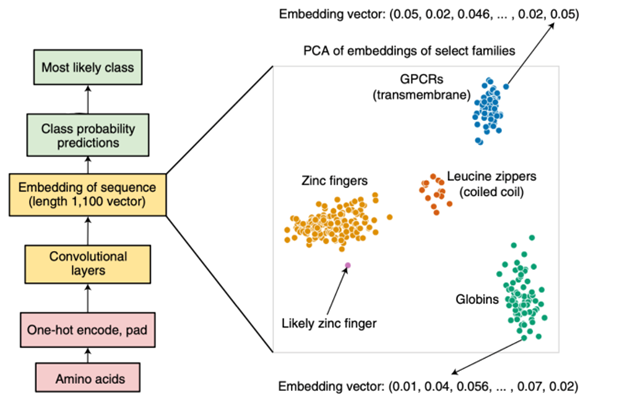
- Curate a dataset of Plasmodium falciparum protein sequences.
- Fine-tune a pre-trained protein language model (ProtT5) on the curated dataset to generate fine-tuned vectorized representations of each protein sequence.
- Refine the embedded representations to ensure that proteins sharing the same pFam annotation cluster closely together within the latent space, while those with different pFam terms remain further apart.
- Encode proteins with unknown functions (test dataset) and examine the nearest neighbors for each protein sequence in the fine-tuned lookup table.
- Evaluate if there is consensus in the annotation of the nearest neighbors and if we can "bridge" the annotation over to the unknown function protein.
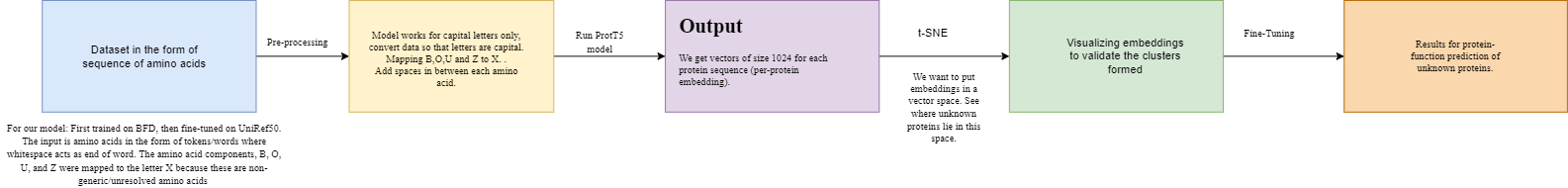
The ProteinT5 model is a pre-trained language model based on the Transformer architecture and trained on a large corpus of protein sequences. It is specifically designed to process protein sequences and extract meaningful representations that capture important features of the proteins, such as their function, structure, and interactions.
The model was trained using a supervised learning approach on a massive amount of protein data, including sequences from a wide range of organisms, such as bacteria, fungi, plants, and animals. The training data was annotated with protein family information, gene ontology terms, and other standardized annotation terms to help the model learn to classify and group proteins based on their functions.
After the training process, the model can be fine-tuned on specific tasks, such as protein classification or annotation of unknown functions, by training it on a smaller dataset specific to the target organism or task. This fine-tuning process allows the model to specialize and improve its performance on the target task.
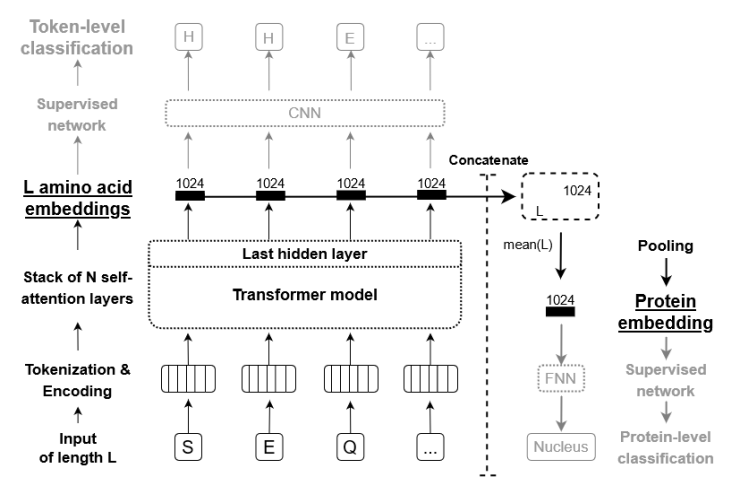
t-SNE (t-distributed Stochastic Neighbor Embedding) is a machine learning technique used for data visualization and dimensionality reduction. It is particularly useful for visualizing high-dimensional data in a low-dimensional space (e.g., 2D or 3D).
The basic idea behind t-SNE is to map high-dimensional data points into a lower-dimensional space, while preserving the pairwise similarities between the points as much as possible. It does this by first computing a probability distribution over pairs of high-dimensional points based on their similarity, and then computing a similar probability distribution over pairs of low-dimensional points. The algorithm then tries to minimize the difference between the two distributions using a gradient descent optimization.
The result is a low-dimensional representation of the data that preserves the pairwise similarities between the original data points, making it easier to visualize and understand the structure of the data. t-SNE is widely used in various fields, such as natural language processing, computer vision, and bioinformatics, for tasks such as document clustering, image recognition, and single-cell RNA sequencing analysis.
We expect to obtain a fine-tuned lookup table that accurately annotates Plasmodium falciparum proteins. This methodology can be used to study specialized features of proteins and perform activities such as locating Transposable Elements.
The purpose of this file is to create a version of the protT5 protein language model on a pfam classification task. This will learn to classify proteins into their respective protein family categories. We'll use the embeddings in this fine-tuned to model to create a vector space. Mapping unknown proteins into this vector space can help predict the function of the unkown protein by judging its similarity to other protein families in the vector space.